AMP®-Parkinson's Disease Progression Prediction¶
Danh sách thành viên:
- 19120454 - Bùi Quang Bảo (Nhóm trưởng)
- 19120462 - Lục Minh Bửu
- 19120151 - Nguyễn Trí Tuệ
- 19120186 - Đỗ Lê Khánh Đăng
Table of Contents: Phía bên phải của trang Kaggle
1. GIỚI THIỆU ĐỀ TÀI 🧠¶
AMP®-Parkinson's
Disease Progression Prediction.
Bệnh Parkinson là một loại bệnh thoái hóa chậm tiến triển của hệ thần kinh trung ương, chủ yếu ảnh hưởng
đến hệ thống vận động. Hiện tại không có thuốc chữa cho bệnh này và bệnh nặng lên theo thời gian 💀.
Thang điểm phân độ bệnh Parkinson của Hiệp hội Rối loạn Vận động (MDS-UPDRS) là một đánh
giá toàn diện về cả triệu chứng chuyển động và phi chuyển động. Đây là thang đo giúp đánh giá tiến triển
của bệnh Parkinson.
Mục tiêu chính:
Phát triển một mô hình được huấn luyện trên tập dữ liệu về mức độ protein và peptide theo thời gian ở những người mắc bệnh Parkinson so với những người cùng tuổi bình thường để dự đoán điểm MDS-UPDR (điểm này dùng cho đo lường sự tiến triển của bệnh nhân mắc bệnh Parkinson).
2. TIỀN XỬ LÝ - KHÁM PHÁ DỮ LIỆU 🔭¶
2.1 Giới thiệu bộ dữ liệu 🗂️¶
# This Python 3 environment comes with many helpful analytics libraries installed
# It is defined by the kaggle/python Docker image: https://github.com/kaggle/docker-python
# For example, here's several helpful packages to load
import numpy as np # linear algebra
import pandas as pd # data processing, CSV file I/O (e.g. pd.read_csv)
import plotly.express as px #visualization
import plotly.graph_objects as go #visualization
import amp_pd_peptide #TEST-API
import matplotlib.pyplot as plt
import seaborn as sns
from collections import Counter
from plotly.subplots import make_subplots
# Input data files are available in the read-only "../input/" directory
# For example, running this (by clicking run or pressing Shift+Enter) will list all files under the input directory
import os
for dirname, _, filenames in os.walk('/kaggle/input'):
for filename in filenames:
print(os.path.join(dirname, filename))
# You can write up to 20GB to the current directory (/kaggle/working/) that gets preserved as output when you create a version using "Save & Run All"
# You can also write temporary files to /kaggle/temp/, but they won't be saved outside of the current session
data_proteins = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_proteins.csv')
data_clinical = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_clinical_data.csv')
data_peptides = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_peptides.csv')
data_supplemental = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/supplemental_clinical_data.csv')
data_proteins
data_clinical
data_peptides
data_supplemental
Kích thước các tập dữ liệu¶
data_peptides.shape
Dữ liệu ở cấp độ peptide - peptide là các tiểu đơn vị thành phần của protein.(train_peptides.csv)
Dữ liệu gồm 6 thuộc tính và 981834 mẫu :
| stt | Tên thuộc tính | Kiểu dữ liệu | Ý nghĩa |
|---|---|---|---|
| 0 | visit_id | objec | Mã định danh ghé thăm khám dịch vụ ( có thể trùng ), gồm các kí tự kiểu chuỗi được nối nhau bằng dấu “_”, là kết hợp giữa 2 thuộc tính patientid visit_month |
| 1 | visit_month | int64 | Tháng thăm khám, so với lần khám đầu tiên của bệnh nhân |
| 2 | patient_id | int64 | Mã ID của bệnh nhân ( Mỗi bệnh nhân có mã ID khác nhau |
| 3 | UniProt | object | Mã ID UniProt cho protein liên quan. Thường có một số peptide trên mỗi protein(Gồm 6 kí tự ). |
| 4 | Peptide | object | Thứ tự các Axit amin xuất hiện trong peptit |
| 5 | PeptideAbundance | float64 | Tần số xuất hiện của axit amin trong mẫu |
data_proteins.shape
Dữ liệu ở cấp độ protein được tổng hợp từ dữ liệu của mức peptide.(train_peptides.csv)
Dữ liệu gồm 232741 mẫu và 5 thuộc tính
| stt | Tên thuộc tính | Kiểu dữ liệu | Ý nghĩa |
|---|---|---|---|
| 0 | visit_id | object | Mã định danh ghé thăm khám dịch vụ ( có thể trùng ), gồm các kí tự kiểu chuỗi được nối nhau bằng dấu “_”, là kết hợp giữa 2 thuộc tính patientid visit_month |
| 1 | visit_month | int64 | Tháng thăm khám, so với lần khám đầu tiên của bệnh nhân |
| 2 | patient_id | int64 | Mã ID của bệnh nhân ( Mỗi bệnh nhân có mã ID khác nhau) |
| 3 | UniProt | object | Mã ID UniProt cho protein liên quan. Thường có một số peptide trên mỗi protein(Gồm 6 kí tự ). |
| 4 | NPX | float64 | Tần suất xuất hiện protein trong mẫu thuộc khoảng [8.460820e+01, 6.138510e+08] |
data_clinical.shape
Thông tin tập dữ liệu clinical khám bệnh lâm sàn về Parkinson
Dữ liệu gồm 2615 mẫu và 8 thuộc tính
| stt | Tên thuộc tính | Kiểu dữ liệu | Ý nghĩa |
|---|---|---|---|
| 0 | visit_id | object | Mã định danh ghé thăm khám dịch vụ ( có thể trùng ), gồm các kí tự kiểu chuỗi được nối nhau bằng dấu “_”, là kết hợp giữa 2 thuộc tính patientid visit_month |
| 1 | visit_month | int64 | Tháng thăm khám, so với lần khám đầu tiên của bệnh nhân |
| 2 | patient_id | int64 | Mã ID của bệnh nhân ( Mỗi bệnh nhân có mã ID khác nhau) |
| 3 | updrs_[1-4] | Float64 | Điểm đánh giá người bệnh cho các mục updrs ,điểm này được so sánh với Unified Parkinson's Disease Rating Scale. Con số cao hơn cho thấy các triệu chứng nghiêm trọng hơn. Mỗi updrs bao gồm một loại triệu chứng riêng biệt updrs 1 thể hiện tâm trạng và hành vi,updrs 2 thể hiện hoạt động thường ngày ,updrs 3 và các chức năng vận động , updrs 4 là điểm đánh giá cho biến chứng của điều trị |
| 4 | upd23b_clinical_state_on_medication | object | Ghi nhận Bệnh nhân có dùng thuốc Levodopa hay không trong quá trình đánh giá UPDRS.[On, Off] Ảnh hưởng chủ yếu đến điểm số của updrs 3 (chức năng vận động). Những loại thuốc này hết tác dụng khá nhanh (theo thứ tự trong một ngày), vì vậy thông thường bệnh nhân sẽ thực hiện kiểm tra chức năng vận động hai lần trong một tháng, cả khi có và không dùng thuốc. |
data_supplemental.shape
Thông tin tập dữ liệu supplemental khám bệnh lâm sàn về Parkinson
Dữ liệu gồm 2223 mẫu và 8 thuộc tính
| stt | Tên thuộc tính | Kiểu dữ liệu | Ý nghĩa |
|---|---|---|---|
| 0 | visit_id | object | Mã định danh ghé thăm khám dịch vụ ( có thể trùng ), gồm các kí tự kiểu chuỗi được nối nhau bằng dấu “_”, là kết hợp giữa 2 thuộc tính patientid visit_month |
| 1 | visit_month | int64 | Tháng thăm khám, so với lần khám đầu tiên của bệnh nhân |
| 2 | patient_id | int64 | Mã ID của bệnh nhân ( Mỗi bệnh nhân có mã ID khác nhau) |
| 3 | updrs_[1-4] | Float64 | Điểm đánh giá người bệnh cho các mục updrs ,điểm này được so sánh với Unified Parkinson's Disease Rating Scale. Con số cao hơn cho thấy các triệu chứng nghiêm trọng hơn. Mỗi updrs bao gồm một loại triệu chứng riêng biệt updrs 1 thể hiện tâm trạng và hành vi,updrs 2 thể hiện hoạt động thường ngày ,updrs 3 và các chức năng vận động , updrs 4 là điểm đánh giá cho biến chứng của điều trị |
| 4 | upd23b_clinical_state_on_medication | object | Ghi nhận Bệnh nhân có dùng thuốc Levodopa hay không trong quá trình đánh giá UPDRS.[On, Off] Ảnh hưởng chủ yếu đến điểm số của updrs 3 (chức năng vận động). Những loại thuốc này hết tác dụng khá nhanh (theo thứ tự trong một ngày), vì vậy thông thường bệnh nhân sẽ thực hiện kiểm tra chức năng vận động hai lần trong một tháng, cả khi có và không dùng thuốc. |
2.2 Tiền xử lý dữ liệu 🗂️¶
2.2.1 Phân tích dữ liệu bị thiếu¶
data_list = [data_proteins, data_peptides, data_clinical, data_supplemental]
data_name = ['data_proteins', 'data_peptides', 'data_clinical', 'data_supplemental']
for i, temp_df in enumerate(data_list):
print(f'Dataframe "{data_name[i]}":\n{temp_df.isnull().sum(axis=0)}\n')
Nhận xét:
- Dữ liệu
proteinsvàpeptideskhông chứa giá trịnull - Dữ liệu
clinicalvàsupplementalcó chứa các giá trịnull
import matplotlib.pyplot as plt
import missingno as msno # Thư viện missingno hỗ trợ trực quan hoá dữ liệu bị thiếu
2.2.1.1. Tập dữ liệu clinical¶
data_clinical_temp = data_clinical
data_clinical_temp['null_values_count'] = data_clinical.isnull().sum(axis=1)
data_clinical_null_per_row = data_clinical_temp['null_values_count'].value_counts()[:-1].reindex([0, 1, 2, 3])
fig = plt.figure(figsize=(8, 8))
plt.pie(
data_clinical_null_per_row,
labels = [str(e) + ' null(s)' for e in data_clinical_null_per_row.index],
colors = ['PaleTurquoise', 'Moccasin', 'LightSalmon', 'Tomato'],
explode = [0.05, 0, 0, 0],
autopct='%1.1f%%'
)
plt.title('Clinical Data - Null values per row')
plt.legend()
plt.show()

Nhận xét:
- Dữ liệu có 51.8% dòng có chứa giá trị null, trong đó:
- 12.7% dòng chứa 1 giá trị null
- 38.8% dòng chứa 2 giá trị null
- 0.4% dòng chứa 3 giá trị null
null_count_labels = [data_clinical_temp[(data_clinical_temp['null_values_count'] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [data_clinical_temp[(data_clinical_temp['null_values_count'] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=3, figsize=(15, 4))
fig.suptitle("Clinical Data - Null values count")
colors = ['Moccasin', 'LightSalmon', 'Tomato']
for i in range(0, 3):
ax = axs[i]
labels = null_count_labels[i]
sns.barplot(x=labels, y=null_count_values[i], ax=ax, color=colors[i])
ax.set_title(f"Rows with {i + 1} null(s)")
ax.set_ylabel("Null values count" if i == 0 else "")
ax.set_xticks([u for u in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")

Nhận xét:
- Với những dòng chứa 1 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupd23b_clinical_state_on_medication - Với những dòng chứa 2 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupdrs_4vàupd23b_clinical_state_on_medication - Với những dòng chứa 3 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupdrs_3,updrs_4vàupd23b_clinical_state_on_medication
msno.matrix(data_clinical, figsize=(10, 5), fontsize=10)
plt.title('Clinical Data - Missing Data Matrix')
plt.show()

Nhận xét:
- Dữ liệu bị thiếu chủ yếu tập trung tại 2 thuộc tính là
updrs_4vàupd23b_clinical_state_on_medication - Dữ liệu bị thiếu tại 2 thuộc tính
updrs_4vàupd23b_clinical_state_on_medicationcó thể có pattern xuất hiện cùng nhau
msno.heatmap(data_clinical, figsize=(10, 5), fontsize=10)
plt.title('Clinical Data - Missing Data Relationship')
plt.show()

Nhận xét:
- 2 thuộc tính
updrs_4vàupd23b_clinical_state_on_medicationcó dữ liệu bị thiếu xuất hiện cùng nhau (Tương quan bị thiếu rất cao, 0.8) - Chúng ta sẽ không kết luận 2 thuộc tính
updrs_1vàupdrs_2có dữ liệu bị thiếu xuất hiện cùng nhau, vì dữ liệu bị thiếu tại 2 thuộc tính này xuất hiện rất ít (chỉ 1 đến 2 mẫu)
2.2.1.2. Tập dữ liệu supplemental:¶
data_supplemental_temp = data_supplemental
data_supplemental_temp['null_values_count'] = data_supplemental.isnull().sum(axis=1)
data_supplemental_null_per_row = data_supplemental_temp['null_values_count'].value_counts()[:-1].reindex([0, 1, 2, 3, 4])
fig = plt.figure(figsize=(8, 8))
plt.pie(
data_supplemental_null_per_row,
labels = [str(e) + ' null(s)' for e in data_supplemental_null_per_row.index],
colors = ['PaleTurquoise', 'Moccasin', 'LightSalmon', 'Tomato', 'Crimson'],
explode = [0.05, 0, 0, 0, 0],
autopct='%1.1f%%'
)
plt.title('Supplemental Data - Null values per row')
plt.legend()
plt.show()

Nhận xét:
- Dữ liệu có 63.2% dòng có chứa giá trị null, trong đó:
- 34.4% dòng chứa 1 giá trị null
- 19.2% dòng chứa 2 giá trị null
- 0.8% dòng chứa 3 giá trị null
- 8.9% dòng chứa 4 giá trị null
null_count_labels = [data_supplemental_temp[(data_supplemental_temp['null_values_count'] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [data_supplemental_temp[(data_supplemental_temp['null_values_count'] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 4))
fig.suptitle("Supplemental Data - Null values count")
colors = ['Moccasin', 'LightSalmon', 'Tomato', 'Crimson']
for i in range(0, 4):
ax = axs[i]
labels = null_count_labels[i]
sns.barplot(x=labels, y=null_count_values[i], ax=ax, color=colors[i])
ax.set_title(f"Rows with {i + 1} null(s)")
ax.set_ylabel("Null values count" if i == 0 else "")
ax.set_xticks([u for u in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")

Nhận xét:
- Với những dòng chứa 1 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupd23b_clinical_state_on_medication, tiếp sau đó làupdrs_4 - Với những dòng chứa 2 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupdrs_4vàupd23b_clinical_state_on_medication - Với những dòng chứa 3 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupdrs_1,updrs_2vàupd23b_clinical_state_on_medication - Với những dòng chứa 4 giá trị
null, dữ liệu thiếu chủ yếu tập trung vào cộtupdrs_1,updrs_2,updrs_4vàupd23b_clinical_state_on_medication
msno.matrix(data_supplemental, figsize=(10, 5), fontsize=10)
plt.title('Supplemental Data - Missing Data Matrix')
plt.show()

Nhận xét:
- Dữ liệu bị thiếu chủ yếu tập trung tại 2 thuộc tính là
updrs_4vàupd23b_clinical_state_on_medication, tiếp theo đó là 2 thuộc tínhupdrs_1vàupdrs_2. - Khác với dữ liệu
clinical, ở dữ liệusupplemental, dữ liệu bị thiếu tại 2 thuộc tínhupdrs_4vàupd23b_clinical_state_on_medicationcó vẻ như không có pattern xuất hiện cùng nhau - Dữ liệu bị thiếu tại 2 thuộc tính
updrs_1vàupdrs_2có thể có pattern xuất hiện cùng nhau
msno.heatmap(data_supplemental, figsize=(10, 5), fontsize=10)
plt.title('Supplemental Data - Missing Data Relationship')
plt.show()

Nhận xét:
- 2 thuộc tính
updrs_1vàupdrs_2có dữ liệu bị thiếu xuất hiện cùng nhau (Tương quan bị thiếu tối đa, 1.0) -> Hễupdrs_1thiếu thìupdrs_2sẽ thiếu - Các cặp thuộc tính có mối tương quan thấp khi bị thiếu dữ liệu:
updrs_1vàupdrs_4: 0.3updrs_1vàupd23b_clinical_state_on_medication: 0.3updrs_2vàupdrs_4: 0.3updrs_2vàupd23b_clinical_state_on_medication: 0.3updrs_4vàupd23b_clinical_state_on_medication: 0.3
2.2.1.3. Kết luận và hướng giải quyết¶
Kết luận chung:
- Trong 4 tập dữ liệu:
- 2 tập dữ liệu liên quan đến hàm lượng protein của bệnh nhân là
proteinsvàpeptideskhông chứa giá trịnull - 2 tập dữ liệu liên quan đến điểm đánh giá UPDRS và tình trạng uống thuốc của bệnh nhân là
clinicalvàsupplementalcó chứa giá trịnull
- 2 tập dữ liệu liên quan đến hàm lượng protein của bệnh nhân là
- Tập dữ liệu
clinical:- Có 51.8% dòng dữ liệu có chứa giá trị
null-> lượng dữ liệu bị thiếu rất lớn - Dữ liệu bị thiếu chủ yếu tập trung tại 2 thuộc tính là
updrs_4vàupd23b_clinical_state_on_medication- Cặp 2 thuộc tính
updrs_4vàupd23b_clinical_state_on_medicationcó tương quan bị thiếu rất cao (0.8).
- Cặp 2 thuộc tính
- Có 51.8% dòng dữ liệu có chứa giá trị
- Tập dữ liệu
supplemental:- Có 63.2% dòng dữ liệu có chứa giá trị
null-> lượng dữ liệu bị thiếu rất lớn - Dữ liệu bị thiếu chủ yếu tập trung tại 2 thuộc tính là
updrs_4vàupd23b_clinical_state_on_medication, tiếp theo đó là 2 thuộc tínhupdrs_1vàupdrs_2.- Cặp 2 thuộc tính
updrs_4vàupd23b_clinical_state_on_medicationcó tương quan bị thiếu không cao như ở tập dữ liệuclinical(0.3). - Cặp 2 thuộc tính
updrs_1vàupdrs_2có tương quan bị thiếu rất cao (1.0) -> Hễupdrs_1thiếu thìupdrs_2sẽ thiếu
- Cặp 2 thuộc tính
- Có 63.2% dòng dữ liệu có chứa giá trị
Phân tích và đề xuất hướng giải quyết:
- Đối với dữ liệu UPDRS, cụ thể gồm
updrs_1,updrs_2,updrs_3vàupdrs_4:- Các cột này là điểm đánh giá người bệnh thông qua các triệu chứng trong các mục của UPDRS, bao
gồm:
updrs_1: đánh giá tâm trạng và hành viupdrs_2: đánh giá các hoạt động thường ngàyupdrs_3: đánh giá các chức năng vận độngupdrs_4: đánh giá các biến chứng của điều trị
- Quan trọng: Điểm số đánh giá cao hơn cho thấy các triệu chứng tương ứng nghiêm trọng hơn. Và điểm
số bằng
0có nghĩa là người bệnh đã bình thường (giá trị0có ý nghĩa). Vì thế mà không thể xem giá trịnulllà giá trị0được. - Nguyên nhân dẫn đến thiếu dữ liệu (giả thuyết):
- Quá trình thu thập dữ liệu đã không thu thập được / quên thu thập điểm kiểm tra đánh giá UPDRS ở một số người bệnh.
- Bệnh nhân không tiến hành kiểm tra đánh giá UPDRS -> không có dữ liệu để thu thập.
- Người bệnh từ chối cung cấp thông tin kiểm tra đánh giá UPDRS
- Các hướng giải quyết:
- Loại bỏ các dòng chứa giá trị
nullở các cột đánh giá UPDRS, hoặc - Gán giá trị
nullở các cột đánh giá UPDRS bằng một giá trị cụ thể, điều kiện là khi nhìn vào giá trị đó, chúng ta có thể biết được rằng giá trị này đã từng lànull, ví dụ: giá trị-1.
- Loại bỏ các dòng chứa giá trị
- Các cột này là điểm đánh giá người bệnh thông qua các triệu chứng trong các mục của UPDRS, bao
gồm:
- Đối với dữ liệu của cột
upd23b_clinical_state_on_medication:- Cột này ghi nhận bệnh nhân có dùng thuốc Levodopa hay không trong quá trình đánh giá và chỉ nhận 1
trong 2 giá trị
OnhoặcOff. Giá trịnullxuất hiện ở cột này không chắc chắn là bệnh nhân có dùng thuốc hay không. - Nguyên nhân dẫn đến thiếu dữ liệu (giả thuyết):
- Quá trình thu thập dữ liệu đã không thu thập được / quên thu thập trạng thái uống thuốc ở một số người bệnh.
- Người bệnh từ chối cung cấp thông tin uống thuốc.
- Các hướng giải quyết:
- Loại bỏ cột
upd23b_clinical_state_on_medication, hoặc - Loại bỏ các dòng chứa giá trị
nullở cộtupd23b_clinical_state_on_medication, hoặc - Gán giá trị
nullở cộtupd23b_clinical_state_on_medicationbằng giá trịUnknown, có ý nghĩa là không biết được tình trạng uống thuốc của người bệnh. Như vậy, cộtupd23b_clinical_state_on_medicationsẽ có 3 giá trị làOn,OffvàUnknown.
- Loại bỏ cột
- Cột này ghi nhận bệnh nhân có dùng thuốc Levodopa hay không trong quá trình đánh giá và chỉ nhận 1
trong 2 giá trị
2.2.2 Kiểm tra dữ liệu bị trùng¶
for i, temp_df in enumerate(data_list):
print(f'Dataframe "{data_name[i]}" contains {temp_df.duplicated().sum()} duplicated samples.')
Nhận xét:
- 4 tập dữ liệu bao gồm
proteins,peptides,clinicalvàsupplementalkhông chứa các mẫu bị lặp.
Kết luận chung:
- Dữ liệu không bị trùng lặp.
2.2.3 Kiểm tra và xử lý outlier¶
2.2.3.1 Tập dữ liệu data_supplement¶
data_supplemental.head()
data_supplemental.shape
Ta không xét cột visit_id vì đây là cột ghép từ patient_id và visit_month. Nên ta sẽ xem xét 2 cột còn lại thay vì cột này.
Cột visit_month: tháng khám bệnh
print(data_supplemental.visit_month.nunique())
data_supplemental.visit_month.unique()
Có 8 mốc thời gian khám bệnh
data_supplemental.visit_month.value_counts().sort_index().plot(kind='bar')
plt.xlabel('Tháng')
plt.ylabel('Số lượng')
plt.title('Số lượng mẫu theo visit_month')
plt.show()

Số lượng dữ liệu ở các mốc cũng không có nhiều bất thường, chỉ có mốc 0 là cao hơn hẵn các mốc còn lại. Có thể hiểu rằng vì đây là tháng đầu tiên các bệnh nhân đến khám bệnh. Nên việc nó cao nhất là điều có khả năng xảy ra.
Cột patient_id: mã bệnh nhân
data_supplemental.patient_id.nunique()
data_supplemental.patient_id.value_counts().plot(kind='box')
plt.ylabel('Số lần khám của 1 bệnh nhân')
plt.title('Box plot số lần khám của 1 bệnh nhân')
plt.show()

Cột updrs_1: điểm UPDRS Part I - Non-Motor Aspects of Experiences of Daily Living (nM-EDL)
data_supplemental.updrs_1.plot(kind='hist')
plt.xlabel('Điểm UPDRS-1')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_1')
plt.show()

Theo bài báo https://www.movementdisorders.org/MDS-Files1/PDFs/Rating-Scales/MDS-UPDRS_Vol23_Issue15_2008.pdf được đính kèm trong mô tả dữ liệu, Part I này sẽ có 13 chỉ tiêu, điểm số mỗi chỉ tiêu là 0 - 4. Nên khoảng giá trị hợp lệ của part 1 là 0 - 52.
Ta thấy toàn bộ giá trị trong dữ liệu đều nằm trong khoảng này.
Ta thấy dữ liệu đang bị lệch phải, và giá trị ở khoảng ~ 25 khá ít. Nên sẽ chuẩn hóa dữ liệu và dùng thống kê để kiểm tra xem đây có phải là outlier hay không.
norm = np.log(data_supplemental['updrs_1']+1)
norm.plot(kind='box')
plt.title('Box plot updrs_1 đã chuẩn hóa log')
plt.ylabel('Giá trị')
plt.show()

Ta thấy đây không phải là outlier
Cột updrs_2: điểm UPDRS Part II - Motor Aspects of Experiences of Daily Living (M-EDL)
data_supplemental.updrs_2.plot(kind='hist')
plt.xlabel('Điểm UPDRS-2')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_2')
plt.show()

Tương tự như trên, Part II có khoảng hợp lệ là 0 - 52 và tất cả giá trị đều nằm trong khoảng này
Tuy nhiên, khoảng giá trị >25 có khá ít mẫu, nên ta sẽ detect outlier như updrs-1
norm = np.log(data_supplemental['updrs_2']+1)
norm.plot(kind='box')
plt.title('Box plot updrs_2 đã chuẩn hóa log')
plt.ylabel('Giá trị')
plt.show()

Ta thấy trong cột này cũng không có outlier
Cột updrs_3: điểm UPDRS Part III - Motor Examination
data_supplemental.updrs_3.plot(kind='hist')
plt.xlabel('Điểm UPDRS-3')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố updrs_3')
plt.show()

Part III có khoảng giá trị từ 0 - 132 (33 chỉ tiêu) và tất cả giá trị đều nằm trong khoảng này.
Cột updrs_4: điểm UPDRS Part IV - Motor Complications
data_supplemental.updrs_4.plot(kind='hist')
plt.xlabel('Điểm UPDRS-4')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố updrs_4')
plt.show()

Part IV gồm 6 chỉ tiêu. Khoảng điểm là 0 - 24 nên tất cả giá trị trong dữ liệu đều hợp lệ.
Dữ liệu hiện tại đang bị lệch nặng về điểm 0. Và khoảng từ 8 trở lên có khá ít dữ liệu.
Tuy nhiên thì nó vẫn phù hợp với xu hướng giảm dần của dữ liệu. Dù vậy ta vẫn có thể xem xét bỏ đi để cải
thiện kết quả nếu cần thiết.
Cột upd23b_clinical_state_on_medication
print(data_clinical.upd23b_clinical_state_on_medication.nunique())
data_clinical.upd23b_clinical_state_on_medication.unique()
value_count = data_supplemental.upd23b_clinical_state_on_medication\
.value_counts()
value_count.plot(kind='bar')
for i in range(len(value_count.index)):
plt.text(i, value_count.values[i]+2,str(value_count.values[i]), ha='center')
plt.title('Số lượng mẫu theo tình trạng sử dụng thuốc')
plt.show()

Ta thấy giữa số lượng mẫu On và Off có sự chênh lệch rất lớn, số lượng Off chỉ có 29. Hơn nữa cột này đang bị thiếu gần 50% dữ liệu nên ta có thể xem xét bỏ cột này ở các bước sau.
2.2.3.2 Tập dữ liệu data_clinical¶
data_clinical.head()
data_clinical.shape
Ta không xét cột visit_id vì đây là cột ghép từ patient_id và visit_month. Nên ta sẽ xem xét 2 cột còn lại thay vì cột này.
Cột visit_month: tháng khám bệnh
print(data_clinical.visit_month.nunique())
data_clinical.visit_month.unique()
month_count = data_clinical.visit_month.value_counts().sort_index()
for i in range(len(month_count.index)):
plt.text(i, month_count.values[i]+2,str(month_count.values[i]), ha='center')
month_count.plot(kind='bar')
plt.title('Số lượng mẫu theo visit_month')
plt.xlabel('Tháng')
plt.ylabel('Số lượng mẫu')
plt.plot()

Ta thấy cột này không có gì bất thường, hầu hết các mốc thời gian đều có số lượng mẫu đủ lớn. Mốc cuối cùng tuy có số lượng chỉ 12 mẫu, nhưng đây là một mốc rất xa (108 tháng) so với lần khám đầu nên sẽ có ít mẫu hơn và phù hợp với xu hướng giảm dần theo các mốc.
Cột patient_id: mã bệnh nhân
data_clinical.patient_id.value_counts().plot(kind='box')
plt.ylabel('Số lần khám của 1 bệnh nhân')
plt.title('Box plot số lần khám của 1 bệnh nhân')
plt.show()

Ở đây ta sẽ chú ý đến 2 số min và max. Ta thấy rằng trong dữ liệu, 1 bệnh nhân ít nhất khám tại 3 mốc tháng khác nhau và nhiều nhất là 17 mốc khám. Điều này hoàn toàn phù hợp với mô tả dữ liệu (1 mốc tháng sẽ chỉ lấy duy nhất 1 kết quả, và dữ liệu của ta có 17 mốc tháng). Vậy cột này cũng không có outlier
Cột updrs_1: điểm UPDRS Part I - Non-Motor Aspects of Experiences of Daily Living (nM-EDL)
data_clinical.updrs_1.plot(kind='hist')
plt.xlabel('Điểm UPDRS-1')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_1')
plt.show()

Khoảng giá trị hợp lệ của part 1 là 0 - 52. Ta thấy toàn bộ giá trị trong dữ liệu đều nằm trong khoảng này.
Tuy nhiên điểm ~30 có số lượng rất ít nên ta sẽ xem xét bằng thống kê
norm = np.log(data_clinical['updrs_1']+1)
norm.plot(kind='box')
plt.title('Box plot giá trị updrs_1 đã chuẩn hóa log')
plt.show()

Ta thấy dữ liệu không có bất thường
Cột updrs_2: điểm UPDRS Part II - Motor Aspects of Experiences of Daily Living (M-EDL)
data_clinical.updrs_2.plot(kind='hist')
plt.xlabel('Điểm UPDRS-2')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_2')
plt.show()

Tương tự như trên, Part II có khoảng hợp lệ là 0 - 52 và tất cả giá trị đều nằm trong khoảng này. Và khoảng điểm > 30 số lượng rất ít, tuy nhiên thì nó phù hợp với xu hướng giảm dần của dữ liệu. Dù vậy, ta có thể xem xét bỏ những điểm này đi để cải thiện kết quả.
Cột updrs_3: điểm UPDRS Part III - Motor Examination
data_clinical.updrs_3.plot(kind='hist')
plt.xlabel('Điểm UPDRS-3')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_3')
plt.show()

Part III có khoảng giá trị từ 0 - 132 (33 chỉ tiêu) và tất cả giá trị đều nằm trong khoảng này. Tuy nhiên ta thấy điểm > 75 nằm tách biệt với phần còn lại. Nên ta có thể xem xét bỏ các điểm này ở các bước sau.
Cột updrs_4: điểm UPDRS Part IV - Motor Complications
data_clinical.updrs_4.plot(kind='hist')
plt.xlabel('Điểm UPDRS-4')
plt.ylabel('Số mẫu')
plt.title('Histogram phân bố điểm updrs_4')
plt.show()

Part IV gồm 6 chỉ tiêu. Khoảng điểm là 0 - 24 nên tất cả giá trị trong dữ liệu đều hợp lệ. Và tương tự như ở file supplemental, ta cũng có thể xem xét bỏ đi các điểm > 17.5 để cải thiện kết quả.
Cột upd23b_clinical_state_on_medication
data_clinical.upd23b_clinical_state_on_medication\
.value_counts().sort_values().plot(kind='bar')
plt.title('Số lượng mẫu theo tình trạng sử dụng thuốc')
plt.show()

Ta thấy 2 lớp On và Off không chênh lệch nhau quá nhiều. Nên cột này không có outlier
2.2.3.2 Tập dữ liệu data_peptides¶
data_peptides.head()
data_peptides.shape
Theo mô tả của dữ liệu, ta có sự ràng buộc ở data_clinical và data_peptides rằng tất cả bệnh nhân và tháng thăm khám của data_peptides phải tồn tại trong data_clinical. Nên nếu có điểm nào nằm ngoài clinical, đó sẽ là điểm cần loại bỏ.
Cột patient_id
not_in = data_peptides[~data_peptides.patient_id.isin(data_clinical.patient_id)]
print('Số mẫu không thỏa ràng buộc ở patient_id:', not_in.shape[0])
Cột visit_month
not_in = data_peptides[~data_peptides.visit_month.isin(data_clinical.visit_month)]
print('Số mẫu không thỏa ràng buộc ở visit_month:', not_in.shape[0])
Ta thấy cả 2 cột đều thỏa điều kiện trên.
Cột UniProt
data_peptides.UniProt.nunique()
data_peptides.UniProt.value_counts().describe()
Dữ liệu có nhiều UniProt khác nhau, tuy nhiên UniProt có số lần xuất hiện thấp nhất cũng có đến 489 mẫu, nên cột này hầu như không có outlier
Cột peptide
data_peptides.Peptide.nunique()
data_peptides.Peptide.value_counts().describe()
Tương tự, cột này cũng hầu như không có outlier
Cột PeptideAbundance
data_peptides.PeptideAbundance.plot(kind='hist')
plt.title('Histogram phân bố của PeptideAbundance')
plt.show()

Dữ liệu đang bị lệch nặng về khoảng 0-0.25xe8. Nên ta sẽ chuẩn hóa và quan sát thống kê của dữ liệu
norm = np.log(data_peptides.PeptideAbundance)
norm.plot(kind='box')
plt.title('Box plot PeptideAbundance sau khi chuẩn hóa log')
plt.show()

Quan sát dữ liệu sau khi chuẩn hóa, ta thấy có nhiều điểm được detect là outlier. Tuy nhiên hầu hết các điểm đều xuất hiện khá gần nhau và dày đặc. Chỉ có 2 điểm phía dưới cùng là cách xa phần còn lại. Nên ta có thể xem xét bỏ 2 điểm này đi ở các bước sau.
data_peptides.iloc[norm[norm<3].index]
Đây là 2 điểm cần được xem xét
plt.figure(figsize=(15,7))
plt.subplot(1,2,1)
plt.title('Phân bố PeptideAbundance theo visit_month')
plt.scatter('PeptideAbundance', 'visit_month', data=data_peptides)
plt.xlabel('PeptideAbundance')
plt.ylabel('visit_month')
plt.subplot(1,2,2)
plt.title('Phân bố PeptideAbundance theo patient_id')
plt.scatter('PeptideAbundance', 'patient_id', data=data_peptides)
plt.xlabel('PeptideAbundance')
plt.ylabel('patient_id')
plt.show()

Quan sát PeptideAbundance theo tháng và bệnh nhân, ta thấy có 1 điểm giá trị 1.75xe8 nằm tách biệt hoàn toàn so với phần còn lại, nên ta có thể xem xét điểm này ở các bước sau.
2.2.3.4 Tập dữ liệu data_proteins¶
data_proteins.head()
Patient_id và visit_month ở file này cũng có ràng buộc với data_clinical như trên
Cột patient_id
not_in = data_proteins[~data_proteins.patient_id.isin(data_clinical.patient_id)]
print('Số mẫu không thỏa ràng buộc ở patient_id:', not_in.shape[0])
Cột visit_month
not_in = data_proteins[~data_proteins.visit_month.isin(data_clinical.visit_month)]
print('Số mẫu không thỏa ràng buộc ở visit_month:', not_in.shape[0])
Ta thấy cả 2 đều thỏa ràng buộc
Cột UniProt
data_proteins.UniProt.nunique()
data_proteins.UniProt.value_counts().describe()
Tương tự như UniProt ở data_peptides, tần suất xuất hiện của UniProt thấp nhất đã là 489. Nên cột này khả năng cao không có outlier
Cột NPX
data_proteins.NPX.plot(kind='hist')
plt.title('Histogram phân bố giá trị NPX')
plt.show()

Cột này bị lệch phải nặng, tập trung chủ yếu ở khoảng 0-0.5xe8
np.log(data_proteins.NPX).plot(kind='box')
plt.title('Box plot giá trị NPX sau khi chuẩn hóa log')
plt.show()

Sau khi chuẩn hóa, ta thấy box plot giúp detect rất nhiều điểm là outlier. Nhưng các điểm này lại nằm khá gần nhau, gần khoảng tập trung của dữ liệu và tần suất cũng rất dày đặc. Nên ta sẽ không xem đây là outlier
2.3 Khám phá dữ liệu 🧬¶
2.3.1 Phân tích các chỉ số thống kê và phân phối 📊¶
Phân tích tập dữ liệu ! Kiểm tra có bao nhiêu bệnh nhân thực trong tập dữ liệu
data_peptides['patient_id'].nunique()
NHẬN XÉT:
- Có 248 bệnh nhân khác nhau trong tập dữ liệu
TẬP TRAIN_PEPTIDES¶
data_peptides.info()
data_peptides.describe()
TẬP_PROTEINS¶
data_proteins.info()
data_proteins.describe()
TẬP CLINICAL¶
data_clinical.info()
data_clinical.describe()
TẬP SUPPLEMENTAL¶
data_supplemental.info()
data_supplemental.describe()
TRỰC QUAN HÓA MỘT SỐ THUỘC TÍNH¶
fig, axes = plt.subplots(4,2 , figsize=(20, 14))
fig.suptitle('Tỉ lệ điểm của 4 thuộc tính Updrs[1-4]')
sns.histplot(ax=axes[0, 0],data=data_clinical, x="updrs_1",color = "#FF99CC")
sns.boxplot(ax=axes[0,1],x=data_clinical["updrs_1"],color = "#FF99CC")
sns.histplot(ax=axes[1, 0],data=data_clinical, x="updrs_2",color = "#00CC33")
sns.boxplot(ax=axes[1, 1],x=data_clinical["updrs_2"],color = "#00CC33")
sns.histplot(ax=axes[2, 0],data=data_clinical, x="updrs_3",color = "#888888")
sns.boxplot(ax=axes[2,1],x=data_clinical["updrs_3"],color = "#888888")
sns.histplot(ax=axes[3, 0],data=data_clinical, x="updrs_4",color = "#FFCC33")
sns.boxplot(ax=axes[3,1],x=data_clinical["updrs_4"],color = "#FFCC33")

NHẬN XÉT¶
- Các chỉ số updrs[1-4] : quan trọng trong việc đánh giá bệnh , các biểu đồ trực quan giá trị điểm của các updrs[1-4] cho thấy mức độ phân bố của các điểm này - điều này là tiền đề để xây dựng mô hình cho ra tri thức
- Đối với chỉ số updrs_1 : Điểm của dữ liệu tập trung chủ yếu ở khoảng từ [0-15]
- Đối với chỉ số updrs_2 : Điểm của dữ liệu tập trung chủ yếu ở khoảng từ [0-10] và cao nhất ở 0
- Đối với chỉ số updrs_3 : Điểm của dữ liệu tập trung chủ yếu ở khoảng từ [0-40] và cao nhất ở 0
- Đối với chỉ số updrs_4 : Điểm của dữ liệu khá thưa thớt, tập trung nhiều ở khoảng từ [0-1] và cao nhất ở 0 và có khá nhiều dữ liệu ngoại lệ
Kiểm tra tỉ lệ người dùng thuốc¶
plt.figure(figsize=(10,6))
plt.xlabel('Trạng thái')
plt.ylabel('Số lượng')
# displaying the title
plt.title("Thuộc tính upd23b_clinical_state_on_medicationd")
data_clinical['upd23b_clinical_state_on_medication'].value_counts().sort_values().plot(kind = 'bar',color='#4169E1');

Nhận xét¶
Như vậy có thể thấy ! Đa số bệnh nhân dùng thuốc Levodopa Với tỉ lệ Có sử dụng ( ON ) xấp xỉ 800 Trạng thái Off là gần 500 Mẫu dữ liệu chứa giá trị NaN khá nhiều
=> Vẫn chưa kết luận được gì ! Dữ liệu thiếu là quá nhiều
df = data_clinical.loc[data_clinical['upd23b_clinical_state_on_medication']=='On']
df_off = data_clinical.loc[data_clinical['upd23b_clinical_state_on_medication']=='Off']
Tỉ lệ điểm của 4 thuộc tính Updrs[1-4] dựa trên visit_month với upd23b_clinical_state_on_medication là On¶
pallete = sns.color_palette("tab10", 5)
fig, axes = plt.subplots(2,2, figsize=(20, 10))
fig.suptitle('Tỉ lệ điểm của 4 thuộc tính Updrs[1-4] dựa trên visit_month với upd23b_clinical_state_on_medication là On')
sns.boxplot(ax=axes[0, 0],x=df["visit_month"],y =df["updrs_1"] ,color = pallete[2])
sns.boxplot(ax=axes[0, 1],x=df["visit_month"],y =df["updrs_2"] ,color = pallete[1])
sns.boxplot(ax=axes[1, 0],x=df["visit_month"],y =df["updrs_3"] ,color = pallete[0])
sns.boxplot(ax=axes[1, 1],x=df["visit_month"],y =df["updrs_4"] ,color = pallete[3])

NHẬN XÉT :¶
- Dữ liệu cho thấy ở các thuộc tính updrs_1,2,3 khá tương đồng nhau , dữ liệu phân bố khá đều ở các tháng , tuy nhiên vẫn có các điểm ngoại lai
- Đối với thuộc tính updrs_4 thì giá trị NaN khá nhiều và xuất hiện nhiều các điểm dữ liệu ngoại lệ có thể là ngoại lai , nhiều hơn so với các updrs còn lại
Tỉ lệ điểm của 4 thuộc tính Updrs[1-4] dựa trên visit_month với upd23b_clinical_state_on_medication là Off¶
fig, axes = plt.subplots(2,2, figsize=(20, 10))
fig.suptitle('Tỉ lệ điểm của 4 thuộc tính Updrs[1-4] dựa trên visit_month với upd23b_clinical_state_on_medication là Off')
sns.boxplot(ax=axes[0, 0],x=df_off["visit_month"],y =df_off["updrs_1"] ,color = pallete[2])
sns.boxplot(ax=axes[0, 1],x=df_off["visit_month"],y =df_off["updrs_2"] ,color = pallete[1])
sns.boxplot(ax=axes[1, 0],x=df_off["visit_month"],y =df_off["updrs_3"] ,color = pallete[0])
sns.boxplot(ax=axes[1, 1],x=df_off["visit_month"],y =df_off["updrs_4"] ,color = pallete[3])

NHẬN XÉT :¶
- Dữ liệu cho thấy giữa updrs_1 và updrs_2 khá tương đồng về dữ liệu - vẫn xuất hiện các điểm ngoại lai ở các thuộc tính này
- Dữ liệu của thuộc tính updrs_3 phân bố khá ít ở 2 tháng 6 và 108 - vẫn xuất hiện các điểm ngoại lai
- Đối với updrs_4 Dữ liệu bị thiếu khá nhiều các điểm ngoại lai chiếm phần lớn trong thuộc tính
2.3.2 Phân tích về mối quan hệ giữa các thuộc tính 📊¶
df_corr = data_clinical.corr()
fig = go.Figure()
fig.add_trace(
go.Heatmap(
x = df_corr.columns,
y = df_corr.index,
z = np.array(df_corr),
text=df_corr.values,
texttemplate='%{text:.2f}',
colorscale ='sunset'
)
)
fig.show()
Nhận xét:
- Cả 4 cột điểm đều có độ tương quan thấp đối với cột visit_month
- Nhưng chúng lại có độ tương quan với nhau khá cao. Từ điều này ta có thể biết được các triệu chứng của căn bệnh có liên quan mật thiết với nhau vì đều ảnh hưởng đến các chức năng vận động
Tiếp theo ta sẽ xem xét diễn biến 4 cột điểm theo thời gian như thế nào
Diễn biến của 4 cột điểm theo các tháng
for upd in ["updrs_1","updrs_2","updrs_3","updrs_4"]:
fig = px.scatter(data_clinical, x="visit_month", y=upd, color="upd23b_clinical_state_on_medication", trendline="ols")
fig.show()
Độ tương quan giữa 4 thang điểm với các loại proteins
data_clinical[data_clinical['upd23b_clinical_state_on_medication']=='Off'].corr()
data_clinical[['visit_month','updrs_1']]['visit_month'].unique()
merge_protein_clinical = pd.merge(data_clinical, data_proteins, on=['patient_id','visit_month'])
merge_protein_clinical.columns
columns = ['updrs_1', 'updrs_2',
'updrs_3', 'updrs_4','UniProt', 'NPX']
columns_2 = ['UniProt','updrs_1', 'updrs_2',
'updrs_3', 'updrs_4']
corr_matrix = merge_protein_clinical[columns].groupby('UniProt').corr().reset_index()
corr_matrix = corr_matrix[corr_matrix['level_1']=='NPX'][columns_2].reset_index(drop=True)
corr_matrix = corr_matrix.set_index('UniProt')
for index in range(0,corr_matrix.T.shape[1],15):
fig = px.imshow(corr_matrix.T.iloc[:,index:index+15],text_auto=True,color_continuous_scale ='sunset')
fig.show()
top_20_proteins = []
count_all = []
for col in corr_matrix.columns.tolist():
temp = corr_matrix.nlargest(20, col).index.tolist()
top_20_proteins.append(temp)
count_all.extend(temp)
print(temp)
occu = pd.DataFrame.from_dict(Counter(count_all), orient='index')
occu = occu.sort_values(by=[0],ascending=False)
occu
Như ta có thể thấy, protein P19827 có sự tương quan lớn đến cả 4 thang điểm.
Các proteins P01594, P02748, P01009, P01861, P02679, P04433 chỉ có sự tương quan lớn
đến 3 thang điểm.
Các protein còn lại thì tương quan với 1 hoặc 2 thang điểm.
Tuy nhiên, chúng ta cũng cần xét tới tỷ lệ của chúng trong tập dữ liệu, nếu độ tương quan cao mà tỷ lệ thấp thì cũng không có ý nghĩa gì.
Tỷ lệ các protein trong mẫu CFS của bệnh nhân
percent_protein = {}
for protein in occu.index.tolist():
percent_protein[protein] = data_proteins[data_proteins.UniProt == protein].shape[0]*100 \
/data_clinical.shape[0]
percent_protein = pd.DataFrame.from_dict(percent_protein,orient='index').sort_values(by=[0],ascending=False)
fig = px.bar(percent_protein,orientation='h')
fig.show()
Các protein đều có tỷ lệ % xuất hiện trong các mẫu lớn hơn 23%, nên chúng ta không cần loại bỏ mẫu nào
3. KHAI THÁC DỮ LIỆU¶
3.1. ARIMA + Linear Interpolation¶
3.1.1. Trích chọn và Rút trích đặc trưng¶
Dựa trên cấu trúc, các bộ dữ liệu có thể kết hợp lại dựa trên biểu đồ sau:
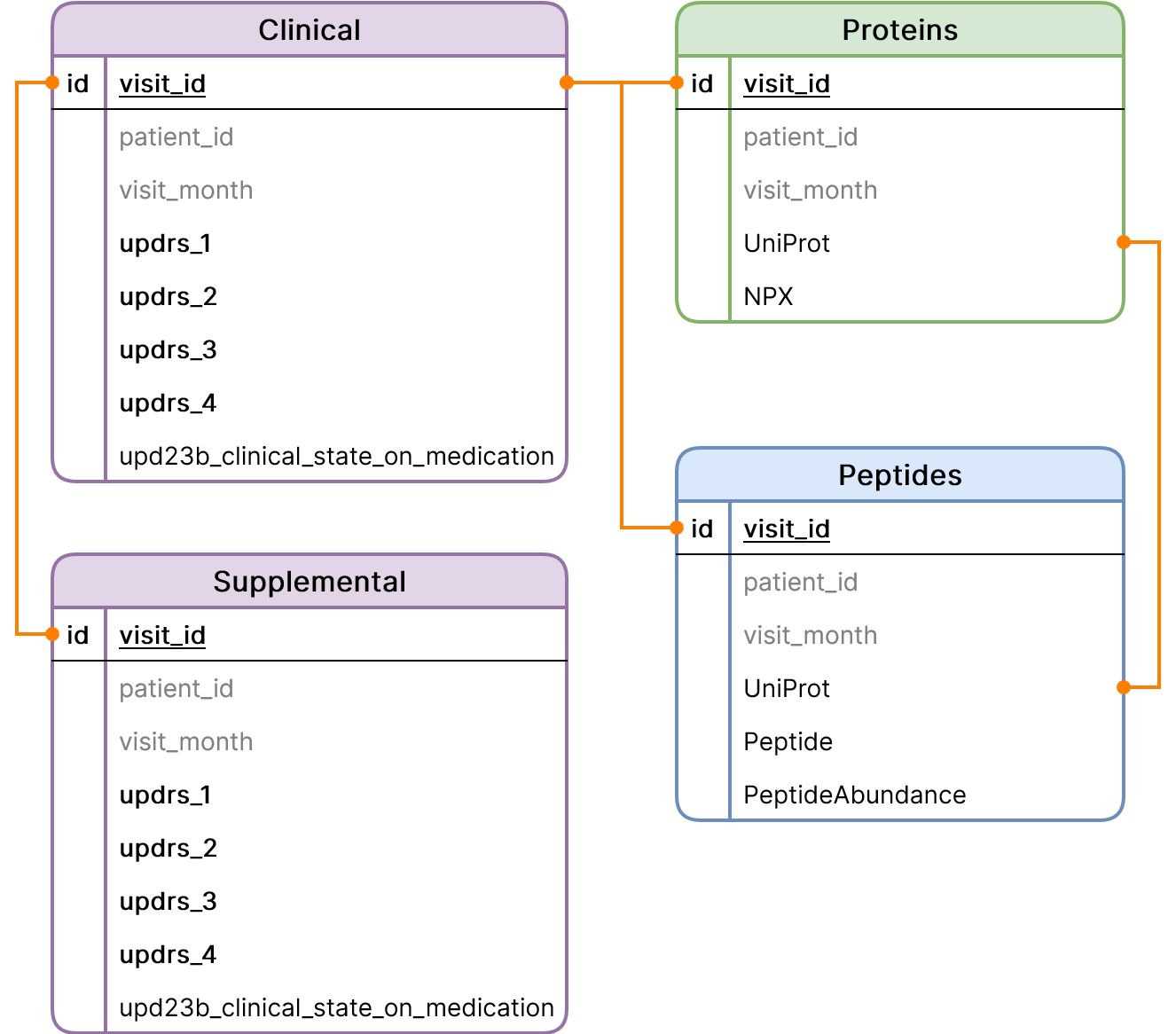
Sau nhiều lần thử nghiệm mô hình với các thay đổi khác nhau, nhóm có nhận xét như sau:
Khi sử dụng mô hình ARIMA:
- Kết hợp bộ dữ liệu
supplementvớiclinicalcho ra kết quả tốt hơn (so với chỉ sử dụng bộ dữ liệuclinical) - Loại bỏ những bệnh nhân khám ít hơn 24 tháng ra khỏi bộ dữ liệu cho ra kết quả tốt hơn.
- Nếu bỏ đi những dòng dữ liệu có
visit_month> 24 (hay nói cách khác, chỉ giữ lại những tháng 0, 6, 12, 24) thì cho ra kết quả tương đương chứ không bị tệ đi. - Nếu 1 bệnh nhân bị thiếu dữ liệu các tháng ở giữa (ví dụ chỉ có dữ liệu tháng 0 và tháng 36, bị thiếu dữ liệu các tháng 6, 12 và 24) thì điền những giá trị thiếu bằng Linear Interpolation cho ra kết quả tốt hơn (so với việc loại bỏ bệnh nhân đó, hoặc là chỉ drop những tháng bị thiếu)
- Nếu không sử dụng Linear Interpolation để điền dữ liệu bị thiếu, thì lúc train mô hình cho điểm UPDRS nào thì mới drop NaN trên cột điểm UPDRS đó thì ra kết quả tốt hơn (so với việc là trước khi train mô hình, drop tất cả những dòng nào có giá trị NaN)
Từ nhận xét trên, ở phiên bản cuối cùng xây dựng mô hình ARIMA, nhóm sẽ thực hiện tiền xử lý như sau:
- Sử dụng kết hợp 2 bộ dữ liệu
clinicalvàsupplement - Loại bỏ những bệnh nhân có thời gian khám ít hơn 24 tháng
- Loại bỏ những dòng dữ liệu có
visit_month> 24 (chỉ giữ lại những tháng 0, 6, 12, 24) - Sử dụng Linear Interpolation để điền giá trị điểm UPDRS bị thiếu
Nhóm sẽ sử dụng các đặc trưng đích cần dự đoán: updrs_1, updrs_2,
updrs_3 và updrs_4 để xây dựng 4 mô hình ARIMA cho 4 chỉ số UPDRS.
import amp_pd_peptide
from matplotlib import pyplot as plt
import pandas as pd
import numpy as np
import missingno as msno
from statsmodels.tsa.arima.model import ARIMA
import statsmodels.api as sm
pd.options.mode.chained_assignment = None # default='warn'
plt.rc("figure",figsize=(12,5))
train_clinical_data = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_clinical_data.csv')
train_clinical_data['source'] = 'standard'
supplemental_clinical_data = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/supplemental_clinical_data.csv')
supplemental_clinical_data['source'] = 'supplemental'
train_clinical_all = pd.concat([train_clinical_data, supplemental_clinical_data])
# train_clinical_all = train_clinical_all[~train_clinical_all.visit_month.isin([3, 5, 9])]
# train_clinical_all = train_clinical_all[train_clinical_all.visit_month.isin([0, 6, 12, 24])]
cli_data = train_clinical_all
pep_data = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_peptides.csv')
pro_data = pd.read_csv('/kaggle/input/amp-parkinsons-disease-progression-prediction/train_proteins.csv')
def get_train_data(pep_data, pro_data, cli_data, join_type = 'inner'):
# Average PeptideAbundance of all proteins
avg_abundance_one_row = pep_data.groupby(['visit_id']).mean().reset_index().drop(columns=['visit_month', 'patient_id']).rename(columns = {'PeptideAbundance':'PeptideAbundance_AVG'})
# Average NPX of all proteins
avg_npx_one_row = pro_data.groupby(['visit_id']).mean().reset_index()[['visit_id', 'NPX']].rename(columns = {'NPX':'NPX_AVG'})
# NPX of important proteins
NPX_P19827 = pro_data[pro_data['UniProt']=='P19827'][['visit_id', 'NPX']].rename(columns = {'NPX':'NPX_P19827'})
# Join on visit_id
train = cli_data.merge(
avg_abundance_one_row, on='visit_id', how=join_type
).merge(
avg_npx_one_row, on='visit_id', how=join_type
).merge(
NPX_P19827, on='visit_id', how=join_type
)
return train
train = get_train_data(pep_data, pro_data, cli_data, join_type = 'outer')
train = train.dropna(subset=['patient_id', 'visit_month'])
train = train.sort_values(by=['patient_id', 'visit_month'], ascending=[True, True])
train
patient_have_visit_than_24 = train[train['visit_month']>=24]['patient_id'].unique()
train = train[train['patient_id'].isin(patient_have_visit_than_24)]
train
visit_month_ls = [0,6,12,24]
patient_id_ls = []
for p in train['patient_id'].unique():
for i in range(len(visit_month_ls)):
patient_id_ls.append(p)
visit_month_ls = visit_month_ls * len(train['patient_id'].unique())
dummy_df = pd.DataFrame(
{'patient_id': patient_id_ls, 'visit_month': visit_month_ls}
)
dummy_df
merged_df = pd.merge(train, dummy_df, on=['patient_id', 'visit_month'], how='outer')
merged_df = merged_df.sort_values(by=['patient_id', 'visit_month'], ascending=[True, True])
merged_df
interpolated_df = merged_df.interpolate(subset=['updrs_1', 'updrs_2', 'updrs_3', 'updrs_4'])
interpolated_df
train = interpolated_df[interpolated_df['visit_month'].isin([0, 6, 12, 24])]
train.index = pd.DatetimeIndex(train['visit_month']).to_period('M')
train
# Seasonal or Non-seasonal? -> Seasonal (period=4)
# Trend or Non-trend? -> Non-trend
# Non-stationary or stationary? -> Stationary
for updrs in ["updrs_1", "updrs_2", "updrs_3", "updrs_4"]:
updrs_values = train[[updrs]].reset_index()[updrs][:200]
res = sm.tsa.seasonal_decompose(updrs_values, period = 4, model="additive")
fig = res.plot()
fig.tight_layout()
plt.show()




Dựa trên cách tiền xử lý mà nhóm đã chọn (cộng với nhiều thử nghiệm với các trường hợp khác nhau), nhóm sẽ tiến hành xây dựng mô hình ARIMA cho các chuỗi dữ liệu có đặc tính:
-
Seasonal (period=4): Tính chu kỳ (chu kỳ = 4)
-
Non-trend: Dữ liệu không có tính xu hướng
-
Stationary: Dữ liệu có tính ổn định
3.1.2. Xây dựng mô hình¶
Mô hình $SARIMA$ (Seasonal AutoRegressive Integrated Moving Average) cấu hình từ 7 tham số:
Với:
3 tham số $p,d,q$ tương tự như ở mô hình $ARIMA$:
-
$p$: Bậc của mô hình $AR$ - autoregressive (số lượng độ trễ thời gian - "lags" của dữ liệu dùng để dự đoán).
-
$d$: Mức độ chênh lệch - regular differencing (số lần mà dữ liệu trừ đi các giá trị trong quá khứ) cần thiết để khiến cho dữ liệu tĩnh (stationary, not trendy). Nếu dữ liệu đầu vào đã tĩnh (stationary) sẵn thì $d=0$.
-
$q$: Bậc của mô hình $MA$ - moving-average (số lượng lỗi - lagged forecast error được đưa vào mô hình).
Mô hình $SARIMA$ bổ sung thêm 3 tham số bắt buộc và 1 tham số tuỳ chọn, có liên quan đến tính chu kỳ (seasonal) như sau:
-
$P$: Tương tự $p$, nhưng mà là bậc của mô hình $SAR$ - seasonal autoregressive.
-
$D$: Tương tự $d$, nhưng là mức độ chênh lệch theo chu kỳ - seasonal differencing.
-
$Q$: Tương tự $q$, nhưng mà là bậc của mô hình $SMA$ - seasonal moving-average.
-
$m$: Số bước/giai đoạn của một chu kỳ thời gian (Ví dụ: Chu kỳ thời gian là 1 năm, chúng ta có dữ liệu của mỗi tháng, vậy $m=12$ vì 1 năm có 12 tháng)
%%time
target = ["updrs_1", "updrs_2", "updrs_3", "updrs_4"]
# Dictionary to store the models for each UPDRS score
model = {u: None for u in target}
for updrs in target:
temp = train #.dropna(subset=[updrs])
y = temp[updrs]
# Fit the ARIMA model
trained = ARIMA(y, order=(4, 0, 1), seasonal_order=(0, 0, 0, 4)).fit()
model[updrs] = trained
model
def get_predictions(my_test, model):
my_test = my_test.fillna(0)
for u in target:
my_test['result_' + str(u)] = 0
# Predict
X = my_test["visit_month"]
if u == 'updrs_4':
my_test['result_' + str(u)] = 0
else:
n = len(model[u].model.endog)
start = n
end = n + len(X) - 1
my_test['result_' + str(u)] = np.ceil(model[u].predict(start=start, end=end).values)
result = pd.DataFrame()
for m in [0, 6, 12, 24]:
for u in [1, 2, 3, 4]:
temp = my_test[["visit_id", "result_updrs_" + str(u)]]
temp["prediction_id"] = temp["visit_id"] + "_updrs_" + str(u) + "_plus_" + str(m) + "_months"
temp["rating"] = temp["result_updrs_" + str(u)]
temp = temp [['prediction_id', 'rating']]
result = result.append(temp)
result = result.drop_duplicates(subset=['prediction_id'])
result['rating'] = result['rating'] + 0.0 # -0.0 -> 0.0
result = result.reset_index().drop(columns=['index'])
return result
%%time
env = amp_pd_peptide.make_env()
iter_test = env.iter_test()
for (test, test_peptides, test_proteins, sample_submission) in iter_test:
my_test = get_train_data(test_peptides, test_proteins, test, join_type='left')
result = get_predictions(my_test,model)
env.predict(result)
result
3.1.3. Kết quả và nhận xét¶
Nhận xét 1¶
Giải thích cho việc:
- Loại bỏ những bệnh nhân có thời gian khám ít hơn 24 tháng -> Kết quả tốt hơn
- Loại bỏ những dòng dữ liệu có visit_month > 24 -> Kết quả tốt hơn
Vì: Khi tiền xử lý dữ liệu như trên, dữ liệu cuối cùng sẽ là 1 chuỗi time-series có cấu trúc
visit_month (thời gian) của 1 bệnh nhân lần lượt là các giá trị 0, 6, 12, 24.
Khi có cấu trúc như thế này, vô hình chung chuỗi time-series sẽ có chu kỳ bằng 4.
Mà mô hình SARIMA lại hoạt động tốt nếu chuỗi dữ liệu có tính chu kỳ, từ đó mà kết quả cho ra tốt hơn.
Nhận xét 2¶
Giải thích cho việc:
- Sử dụng kết hợp 2 bộ dữ liệu clinical và supplement -> Kết quả tốt hơn
Vì:
- Với hướng tiếp cận không sử dụng dữ liệu proteins và peptides, 2 bộ dữ liệu
clinicalvàsupplemental(vốn đã có cấu trúc giống nhau) sẽ mang lại giá trị tương đương nhau (so sánh với trường hợp sử dụng dữ liệu đo nồng độ proteins thì rõ ràng bộ dữ liệusupplementalmang giá trị thấp hơn vì bệnh nhân ở tập này không được đo nồng độ proteins). - Vì giá trị của 2 bộ dữ liệu tương đương nhau nên khi kết hợp, kết quả cho ra tốt hơn (vì đơn giản là mô hình có nhiều dữ liệu hơn để học)
Kết luận¶
- Bệnh Parkinson có xu hướng tiến triển và nặng dần đều theo thời gian. Khi sử dụng Linear Interpolation để điền giá trị điểm UPDRS bị thiếu, mô hình cho ra kết quả tốt hơn giúp củng cố hơn kết luận này.
- Bệnh nhân có sử dụng thuốc trong quá trình điều trị, mặc dù vẫn nặng lên theo thời gian, nhưng tiến triển sẽ chậm hơn so với bệnh nhân không sử dụng thuốc trong điều trị.
- Những proteins có ảnh hưởng lớn đến tiến triển bệnh Parkinson:
- P19827 có sự ảnh hưởng lớn đến cả 4 thang điểm UPDRS
- Các proteins P01594, P02748, P01009, P01861, P02679, P04433 có sự ảnh hưởng lớn đến 3 trên 4 thang điểm.